📜 GraphNVP: An Invertible Flow Model for Generating Molecular Graphs
GraphNVP
In this post we introduce our recent paper “GraphNVP: An Invertible Flow Model for Generating Molecular Graphs”.
Molecule Generation
Discovery of new molecules with desirable phramocological properties is a crucial problem in computational drug discovery. Traditionally, this task is performed by clinically synthesizing candidate chemical compounds and running experiments over them. However, due to the sheer size of chemical space, synthesizing moelcules and extensively performing experiments on them is an extremely time consuming task. Instead of searching through the space of molecules with desirable properties, de novo drug design involves designing new chemical compunds with the properties that we are interested in.
Recent advancements in deep learning, specially deep generative models proved to be invaluable in de novo drug designing.
The choice of molecule representation
An important step in the application of deep learning on molecule generation is how chemical compounds are represented. Earlier models relied on a string-based representation named SMILES. RNN-based language models or Variational Autoencoders (VAE) are used to generate SMILES strings which are then converted to molecules. A major issue in using SMILES strings is that they are not robust to minor changes of a string, resulting in drastically different molecules although the corresponding SMILES strings are almost similar. These problems propmpted recent researches to rely on more expressive graph representations of molecules. Therefore, this problem became to known as molecular graph generation.
A molecule is represented by an undirected graph, in which the atoms and bonds are represented nodes and edges respectively. The structure of a molecule is represented by an adjacency tensor \( A \) and a node feature matrix \( X \) is used to represent the type of atoms (e.g., Oxygen, Fluorine etc.). The molecule generation problem reduces to generation of graphs which can represent valid molecules. This is a problem in which deep generative models such as GANs or VAEs can be leveraged. We can classify previous work into two categories based on how they generate a graph. Some models generate molecular graphs sequentially such that nodes (atoms) and edges (bonds) are added in a step-by-step fashion. The alternative is straightforward, generate a graph in a single step in a similar manner to image generation models.
The importance of reversibility
A significant advantage of the invertible flow-based models is they perform precise likelihood maximization, unlike VAEs or GANs. We believe precise optimization is crucial in molecule generation for drugs as they are highly sensitive to a minor replacement of a single atom (node). An additional advantage of flow models is since they are invertible by design, perfect reconstruction is guaranteed and no time-consuming procedures are needed. Simply running the reverse step of the model on a latent vector results in a molecular graph. Moreover, the lack of an encoder in GAN models makes it challenging to manipulate the sample generation. For example, it is not straightforward to use a GAN model to generate molecules that are similar to a query molecule (e.g., lead optimization for drug discovery), while it is easy for flow-based models.
Our model

GraphNVP, our proposed model is shown above. GraphNVP is the first graph generation model based on the invertible flow which follows one-shot generation strategy. We introduce two latent representations, one for node assignments and another for the adjacency tensor, to capture the unknown distributions of the graph structure and its node assignments respectively. We use two new types of coupling layers: Adjacency Coupling and Node Feature Coupling for obtaining these two latent representations. During graph generation, first we generate an adjacency tensor and then the node feature tensor is generated using graph convolutional networks.
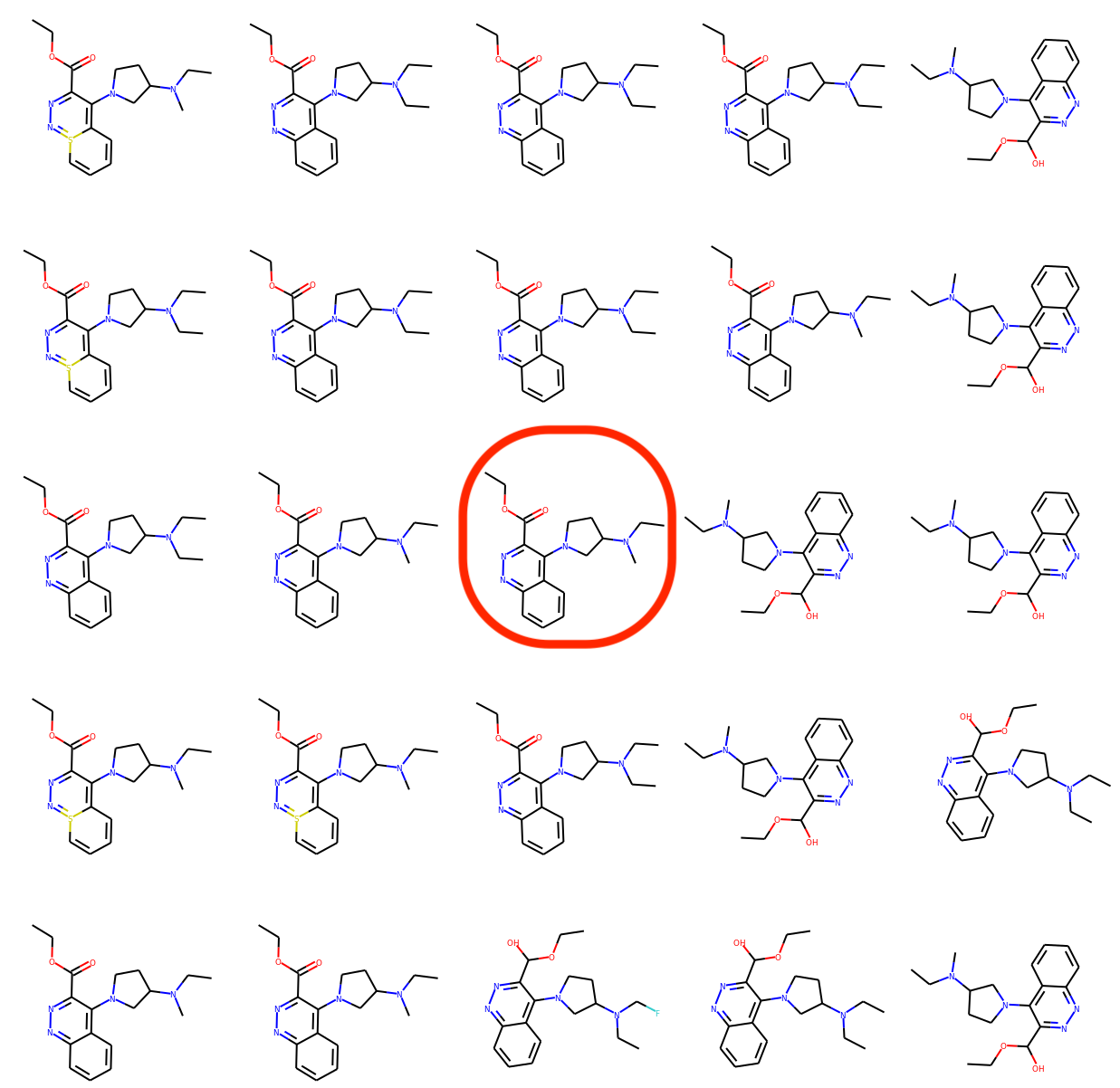
Qualitative results
We randomly select a molecule from the training set and encode it into a latent vector \( z_0 \) using our proposed model. Then we choose two random axes which are orthogonal to each other. We decode latent points lying on a 2-dimensional grid spanned by those two axes and with \( z_0 \) as the origin. The visualization below indicates that the learned latent space is smooth such that neighboring latent points correspond to molecules with minor variations.